1. What is the working principle of a physical adsorption analyzer (specific surface and porosity analyzer)?
Since there is no direct measurement of the surface of the tool, people use the gas molecules of the known molecular cross-section as probes to create conditions for the gas molecules to cover the entire surface of the sample to be tested (adsorption). The number of molecules adsorbed multiplied by the molecular cross-sectional area is considered to be the specific surface area of ​​the sample. Measurement of specific surface area includes all gases that can reach the surface, whether external or internal. Physical adsorption is generally a weak reversible adsorption, so the solid must be cooled to the boiling temperature of the gas and a theoretical method is chosen to calculate the surface area from the single molecule coverage. The specific surface and porosity analysis instrument is such an instrument that creates the corresponding conditions for complex calculations. ?
2. Is the specific surface area value measured?
The specific surface area value is not measured and is calculated. We measure the adsorption isotherm of the sample, and then select the appropriate theoretical model to calculate the specific surface area of ​​the sample based on the characteristics of the sample. Therefore, the specific surface measurement process is actually an analysis process. Because different people may have different knowledge of the sample, experimental data analysis of the same set of adsorption isotherms may report different specific surface area results. Therefore, when "measuring" the surface, keep in mind that this is an "analysis" process. ?22. Is BET a better surface? How many methods are there to calculate the specific surface area?
The BET method is only one theory in the surface analysis method. Langmuir revealed for the first time the nature of adsorption, which is based on the theory of monolayer adsorption and is suitable for sample analysis with only micropores. ?
The BET theory was published in 1938 and its official name is the theory of multi-layer adsorption, which is a modification of the Langmuir theory. BET is the acronym for the three surrogate names of the theory. Since the BET method is suitable for most samples, it is currently the most popular surface analysis method. However, the BET method is not applicable to all samples, so when the microporous material is analyzed by the analysis method of the mesoporous material, the BET specific surface value automatically generated by the physical adsorption analyzer is wrong. Both ISO9277-2010? and IUPAC? specify the BET surface analysis method for microporous materials and the method for determining BET results. The calculation results given by different theoretical models are different, so the theoretical model most suitable for the nature of the sample should be selected according to the assumptions of the theoretical model. Most theoretical models are named according to the inventor's name or abbreviation and can be used to calculate the theoretical model of the specific surface.
Includes Langmuir, BET, BJH, DR and NLDFT. ?? NLDFT is a non-local density functional theory. Studies have shown that NLDFT calculates the closest surface value to the true value, and the theory is applicable to microporous and mesoporous materials.
? 3. What is the principle of measuring specific surface by physical adsorption?
The commonly used adsorbent gas is nitrogen, which has become the standard adsorbent for surface analysis. This is because high-purity nitrogen is readily available; in addition, liquid nitrogen is readily available as the most suitable coolant; third, nitrogen has a greater strength for interaction with most solid surfaces; finally, nitrogen at 77.35K The cross-sectional area is 0.162 nm 2 , and the value that must be used in the BET calculation has been widely accepted. In conventional volumetric techniques, relative pressures less than an integer are achieved by creating partial vacuum conditions. In known fixed volumes, precise high-precision pressure sensors are used to monitor pressure changes caused by the adsorption process. A series of gas adsorptions at different relative pressures need to be measured. Typically, the instrument collects at least 3 data points between 0.025? and 0.30? relative pressure. The experimentally determined data is recorded in pairs of values: the volume of gas at standard temperature and pressure (STP) (VSTP)? indicates the amount of gas adsorption, which corresponds to the relative pressure (P/Po). The graphs drawn from these data are referred to as adsorption isotherms. ?
4. Which important terms should you know at least in physisorption analysis?
In the specific surface area calculation and instrument parameter settings, you should be exposed to the following terms or parameters:
(1) Avogadro constant: 6.022?x?10 23?
(2) BET: This is the abbreviation of three people, they are: S.? Brunauer, ?P.?Emmet? and E.?Teller. They are using
The inventor of the multi-layer gas adsorption theory to calculate the specific surface area. ?
(3) Cross-sectional area (Area): The area occupied by a single adsorbed gas molecule. ?
(4) Molar volume: the volume occupied by one mole of gas. Is it equal to 22,414 cc? (22.414 liters) at standard temperature and pressure?
(5) Molar (dimensionless): The amount of a substance containing an atom or molecule of the Avogadro constant. ?
(6) Monolayer: It is represented by the subscript m, which means a layer of adsorbed gas whose thickness is only a single molecular thickness. ?
(7) Relative pressure P/Po: ratio of absolute pressure P to saturated vapor pressure. Its value is between 0 and 1. ?
(8) Saturated vapor pressure Po: The pressure at which a gas liquefies at a given temperature. ?
(9) Standard temperature and pressure volume: the volume occupied by a certain amount of gas at a standard temperature of 0 ° C (273.15 K)? and a standard atmospheric pressure. ?
5. Why is nitrogen commonly used for surface and pore size analysis? Can you use other gases?
As mentioned earlier, gas molecules are used as adsorption probes to analyze specific surfaces, so it should meet the following application conditions:
1) The gas molecules are relatively inert, ensuring that they do not chemically interact with the adsorbent;
2) In order to adsorb sufficient gas to the solid surface, the solid must be cooled during the measurement, usually to the boiling point of the adsorbed gas, thus requiring the coolant to be relatively easy to obtain;
3) Meet or meet the conditions of use of the ideal gas equation. ?
The adsorption and desorption curves of the gas are measured at a constant low temperature. The gases used are those which form a physical adsorption on the solid surface, especially nitrogen at 77.4 K, argon at 77.4 K or 87.3 K, or 195 K and Carbon dioxide at 273.15K. Because nitrogen is very cheap, it is widely used as an adsorbed substance. Due to the different sizes of gas molecules, the holes that can be entered are also different, so the measurement temperature may be different and the results may be different. ?
Since nitrogen is not a completely inert gas, quadrupole moments can occur with the pore walls. IUPAC officially recommended in 2015 that nitrogen is not suitable for the analysis of microporous samples. Argon gas at 87K should be used as the adsorption gas. ?
6. Why do you use liquid nitrogen for surface and pore size analysis? Don't you have to?
• If nitrogen is used as the adsorbed gas, the solid sample needs to be cooled to the boiling temperature of liquid nitrogen during the analysis (77.35K). Liquid nitrogen is a relatively inexpensive experimental material that is relatively easy to obtain, so we need to use liquid nitrogen to get the temperature required for the sample. However, it should be noted that only pure liquid nitrogen can reach this temperature, and impure liquid nitrogen will cause calculation errors due to high temperature and cannot be used. In addition, liquid nitrogen exposed to the air condenses air, causing a decrease in the purity of liquid nitrogen. Therefore, the liquid nitrogen remaining after the experiment should be discarded, and the liquid nitrogen storage tank cannot be returned to cause the purity of the storage liquid nitrogen to decrease. ?
?? If you do not use liquid nitrogen, we can use mechanical refrigeration to make the sample end at 77.35K. At present, the commercial Cryocooler cryogenic temperature system can set the sample analysis temperature between 20K and 320K, which greatly facilitates the experimental design. ?
7. How to judge the liquid nitrogen is not pure?
Because nitrogen accounts for 78% of the air, its saturated vapor pressure is about atmospheric plus 10. The following conditions indicate that the liquid nitrogen is obviously impure: the ambient atmospheric pressure is 760 mmHg, but the measured nitrogen saturated vapor pressure is greater than 790 mmHg; the liquid nitrogen color is blue, indicating that it contains liquid oxygen; the measured nitrogen saturated vapor pressure It is 750mmHg, but the ambient atmospheric pressure is only 700mmHg, which is obviously higher than the atmospheric pressure at that time; the instrument's liquid level sensor is “failed†and the liquid level is not detected. This means that the liquid level temperature may be higher than the temperature response range set by the sensor;
The linearity during the analysis is very good, but it deviates much from the conventional values. ? 
8. Why should the sample be degassed before performing the sorption analysis?
Solid surfaces must be cleaned of contaminants such as water and oil prior to gas adsorption experiments. In most cases, the surface cleaning (degassing) process is to place a solid sample in a glass sample tube and then heat it under vacuum. The image to the right shows the surface of the solid particles after pretreatment, which contains cracks and pores of different sizes and shapes. ?
9. How to choose the degassing temperature of the sample?
The higher the system temperature, the faster the molecular diffusion motion, so the better the degassing effect. Usually, the degassing station equipped with the instrument can heat up to 400 ° C, but the first principle of selecting the degassing temperature is not to damage the sample structure. In general, the safe degassing temperature of oxides such as alumina and silica can reach 350 ° C; the safe degassing temperature of most carbon materials and calcium carbonate is around 300 ° C; while the hydrate needs to be low. Much more degassing temperature. For organic compounds, pretreatment can also be carried out by a degassing station, but most organic compounds have a lower softening temperature and glass transition temperature and must be confirmed in advance. For example, magnesium stearate commonly used in the medical field, the United States Pharmacopoeia (USP) has a degassing temperature of 40 ° C. If the degassing temperature is set too high, it will cause irreversible changes in the structure of the sample. For example, sintering will reduce the specific surface area of ​​the sample, and decomposition will increase the specific surface area of ​​the sample. However, if the degassing temperature is set too low for insurance, the surface treatment of the sample may be incomplete, resulting in a small analysis result. Therefore, in the case of uncertain degassing temperatures, it is recommended to use a chemical manual such as the?Handbook? of?Chemistry? and?
Physics? (CRC, ?Boca?Raton, ?Florida), and standard methods published by various standards organizations, such as ASTM, as a reference. The degassing temperature should not be chosen higher than the melting point of the solid or the phase transition point of the glass. It is recommended not to exceed half the melting point temperature. Of course, if
Conditions permit, using a thermal analyzer to get the most accurate degassing temperature. In general, the degassing temperature should be the temperature of the platform section on the thermogravimetric curve. ?
10 How to determine the degassing time of the sample?
Corresponding to the degassing temperature is the degassing time. The longer the degassing time, the better the sample pretreatment effect. The choice of degassing time is related to the complexity of the sample well. In general, the more complex the pores, the higher the micropore content and the longer the degassing time; the lower the selected degassing temperature, the longer the degassing time required for the sample. The degassing time can be determined by analyzing the change in the BET result of the sample at the same degassing temperature. If the BET results are the same at different degassing times (2 hours, 4 hours and 6 hours), the degassing time must be chosen to be the shortest; if the change is not large, the compromise should be chosen; if the BET results with the degassing time The extension continues to grow, indicating that the pores are complex, and there are deep-seated adsorbed water molecules due to hydrogen bonding, exposing the blocked pores and area. For general samples, IUPAC recommends a degassing time of no less than 6 hours, while those requiring low temperature degassing require much longer degassing time. For some microporous samples, the degassing time may even be more than 12 hours. However, as a special case, the United States Pharmacopoeia (USP) stipulates that the degassing time of magnesium stearate is only 2 hours. ?
Since the degassing temperature, degassing time, and degassing vacuum are all related to the specific surface area value, it is unavoidable that there is an error in the BET result. Therefore, the sample processing conditions need to be fixed for relative comparison. When comparing with literature values, also pay attention to the sample pretreatment and analysis conditions in the literature. ?
11? When degassing the sample, should you choose vacuum degassing or flow degassing? What are the characteristics of each of the two methods?
Flow degassing is generally used for rapid analysis of specific surfaces. It is very good for removing a large amount of weakly bound adsorbed water on the surface. However, the water adsorbed in the pores can only be diffused to the surface after prolonged purging. Bring out. ?
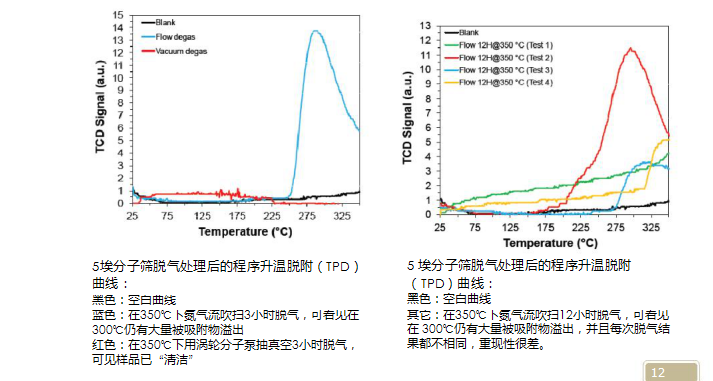
Vacuum degassing is not good for removing a large amount of weakly bound adsorbed water on the surface, because water will diffuse in the pump, causing the pump's pumping force to drop. However, the water adsorbed in the pores does not need to diffuse to the surface for a long time and is then taken out. Therefore, for samples with higher water content, they should be baked in an oven overnight and then placed on a vacuum degassing station to protect the vacuum pump. For vacuum degassing, the cleaning ability of the sample is significantly better than that of the flow degassing (see the figure below), but at the same time, the vacuum degree is different, and the degassing efficiency is significantly different. For ultra-microporous samples, deep-layered adsorbed water molecules can block the pores due to hydrogen bonding. They must be degassed by molecular pump to remove them. The vacuum of the degassing station must reach the same vacuum as the analytical station. Temperature-programmed desorption after degassing of 5 angstrom molecular sieves (TPD)
curve:
12. What are the requirements for degassing a hydrophilic ultra-microporous sample?
? The vacuum degassing method of the molecular pump. In this way, the sample can be degassed in a completely oil-free system. The adsorption measurement often starts at a relative pressure (P?/P 0) of 10-7 microporous material, especially recommended by a low vacuum diaphragm pump plus a turbine.
Degassing of hydrophilic microwell samples is extremely challenging because it is very difficult to remove previously adsorbed water from narrow micropores. Therefore, high temperatures (350 ° C) and long degassing times (usually no less than 8 hours) are required. For some zeolite molecular sieve samples, a special heating procedure is required, that is, at temperatures below 100 ° C, most of the pre-adsorbed water can be slowly removed. The degassing temperature is gradually increased until the final degassing temperature. This is done to avoid damage to the potential structure of the sample due to the influence of surface tension and hydrothermal damage of the vapor. ?
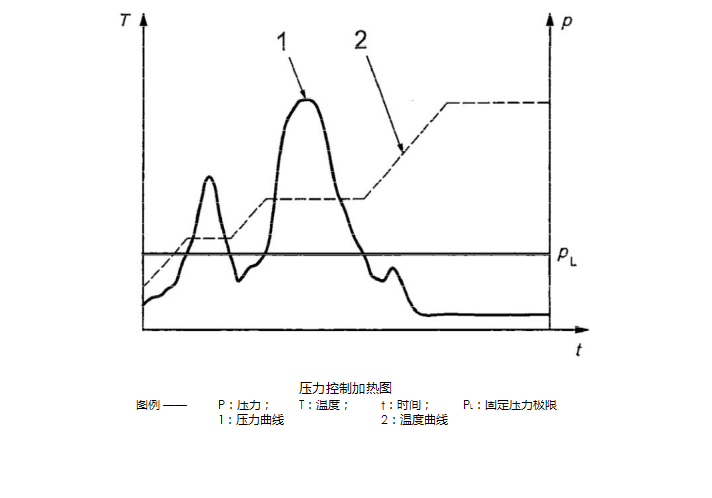
The most recent ISO?9277:2010 standard "Determination of specific surface area for solid gas adsorption? -?BET method" requires: For sensitive samples, pressure-controlled heating is recommended (see figure below). This process involves varying the heating rate with vacuum gas degassing conditions as the gas pressure of the porous material changes. When the material desorbed from the surface of the sample causes the pressure to exceed a fixed limit P (usually about 7 to 10 Pa), the temperature rises and the temperature remains constant until the pressure is below the limit. Then the system continues to heat up. ?
This method is particularly useful for avoiding structural changes in the microporous material because the faster heating rate causes a large amount of adsorbed water to be vaporized centrally, thereby destroying the fragile microporous structure. In addition, the method is very safe to prevent the precipitation of the ultrafine powder material due to the release of water vapor or other vapors in the pores. ?
13. What gas should be backfilled after degassing and unloaded?
?
It is best to use adsorbed gas (nitrogen) as the backfill gas to prevent or minimize the weighing error caused by the buoyancy of the gas. When the sample tube is backfilled with helium, the sample tube will weigh a lot less than the backfill air or nitrogen, with an error of about 1 milligram per milliliter of sample volume. This weighing error is very significant if the sample weight is <50 mg. ?
14. What are the experimental techniques for physical adsorption measurements?
The physical adsorption analysis mainly measures the relationship between the adsorption amount of the sample and the pressure at a certain temperature, that is, the adsorption isotherm curve. The amount of adsorption as a function of pressure can be achieved by volumetric measurement (capacity method) and gravimetric analysis. ?
1) Gravimetric analysis consists of a sensitive microbalance and a pressure sensor that directly measures the amount of adsorption, but requires buoyancy correction (while buoyancy cannot be directly measured). Gravimetric analysis is a convenient method of study when carried out in a relatively small temperature range centered at room temperature. ?
In the gravimetric method, the adsorbate cannot be directly connected to the temperature regulating device, so it is not acceptable at low temperature or high temperature.
Easy to control and measure the true temperature of the adsorbate. Therefore, at liquid nitrogen temperature (77.35K) or liquid argon temperature (87.27K)
The measurement of nitrogen, argon and helium adsorption mainly relies on volume measurement. ?
2) The volumetric measurement method, ie the vacuum capacity method, is based on the calibrated volume and pressure, using the conservation of total gas volume. The amount of adsorption is calculated using the difference between the total amount of gas entering the sample tube and the amount of gas in the free space. Both volumetric and gravimetric methods require that the measured adsorption reaction occur in a static and quasi-equilibrium state. In the quasi-equilibrium state, the adsorbed gas continuously enters the sample tube at a low rate, and the desorption curve is obtained by continuous reduction of pressure. The most difficult part of the relevant quasi-static equilibrium process is that we need to achieve a satisfactory balance of state at all times. In order to detect such an equilibrium state, the analysis should be repeated using a slow gas release rate (gassing). If the same data is obtained at two different gas rates, the correctness of the analysis results can be confirmed. The main advantage of this method is that it can achieve a true equilibrium state and can obtain extremely high resolution adsorption isotherms. ?
3) Continuous flow method: In contrast to the quasi-static equilibrium method, this is a method in which a mixed gas stream of a carrier gas (helium) and an adsorbed gas (e.g., nitrogen) is continuously passed through a fluidized bed in which a sample is placed. The adsorption of nitrogen by the sample causes a change in the composition of the gas. A thermal conductivity detector (TCD) monitors this change and calculates the amount of adsorption. This method is still widely used for rapid analysis of single-site specific surface area. ?
15. What is free space? What is the dead volume? How does it affect measurement sensitivity?
The physical adsorption experiment by the vacuum volume method was carried out in a closed space. The volume of the manifold above the sample tube valve and the volume below the sample tube valve together constitute the system volume required for the static volumetric physical adsorption measurement, in this space of the latter (ie the space of the sample tube), except for the sample The volume occupied, the remaining space is free space (free? space), the volume it occupies is called the dead volume (void? volume). Free space is the area where the adsorbate molecules in the system transmit and diffuse. If the physical adsorption amount of the sample is to be accurately calculated, the dead volume value is the basis for accurate data collection. Because the basis of the vacuum volume method is pressure, the calculation basis of the adsorption amount is the ideal gas state equation. Therefore, the greater the pressure difference of the adsorbate gas during the diffusion process, the more accurate the absolute amount of gas is calculated. The smaller the dead volume of the system, the higher the sensitivity to pressure changes and the more accurate the calculation of the adsorption amount. In other words, under the same conditions, the smaller the dead volume of the system, the higher the instrument measurement accuracy. ?
16. What are the methods for determining the dead volume of free space?
The dead volume should be determined before or after the adsorption isotherm is determined. According to the ISO15901 standard, there are two ways to determine the dead volume.
law:?
1) Measurement method: Volume calibration is performed using helium at the measured temperature. This is the classic dead volume measurement method with the highest accuracy.
The application premise is based on the following two assumptions:
i. Helium is not adsorbed or absorbed by the adsorbent material;
Ii. Helium cannot penetrate into areas where adsorbent substances (such as nitrogen) cannot enter. ?
2)? Calibration curve method: The determination of the dead volume is separated from the adsorption measurement, and the blank test is performed in advance with the adsorption gas tube, and then stored for use (NOVA mode). For example, the volume of the empty sample tube is first determined with nitrogen at ambient temperature, and then a blank experiment is performed with the empty tube under the same experimental conditions as the adsorption measurement (the temperature and relative pressure ranges are the same). The resulting calibration curve essentially represents the detection of multi-point free space. The sample volume is corrected as necessary by inputting the sample density (ie, the skeletal density), or at ambient temperature, before the adsorption analysis begins, the specific gravity is measured with nitrogen (if the adsorption effect of nitrogen at room temperature is negligible). This method is not only suitable for the determination of specific surface area and mesoporous isotherm adsorption line, but also saves helium gas and includes the correction of the adsorption of the pipeline material to the adsorbed gas. The blank curve for a specific sample tube can be used multiple times, so the step of measuring the dead volume with helium is omitted for each sample, which shortens the analysis time and is a method for quickly determining the specific surface or adsorption curve. The IUPAC also noted in its 2015 report that the calibration curve method is advantageous for zeolites and activated carbon adsorbents containing extremely narrow micropores, that is, the NOVA method for determining dead volume is also suitable for zeolite molecular sieves and activated carbon. Microporous analysis, the applicability of MOF materials has also been experimentally supported.
?
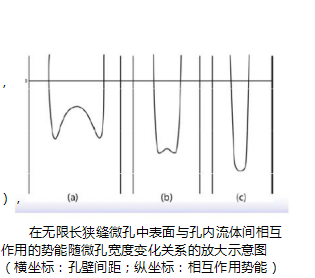
17. What is the relationship between micropore diameter and gas pressure?
In the micropores, the interaction potential energy between the pore walls overlaps each other, so the physical adsorption in the micropores is stronger than the physical adsorption in the wider pores or on the outer surface (see the right figure). Thus, the micropores are sequentially filled at a very low relative pressure (<0.01). That is, the aperture and
The pressure has a corresponding relationship. As the pressure gradually increases from the high vacuum, the gas molecules always fill the smallest hole (c) first, then the larger hole (b), and then the larger hole (a). analogy. Between the surface of the infinitely long slit micropore and the fluid in the hole
An enlarged view of the relationship between the potential energy of the action and the width of the micropore (abscissa: hole wall spacing; ordinate: interaction potential energy)
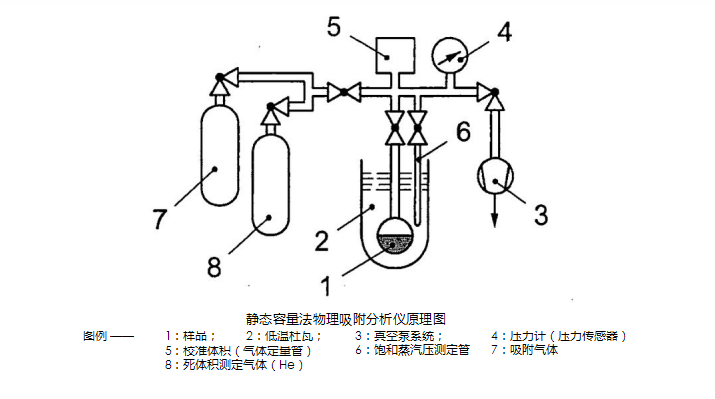
18. What are the components of the static capacity method physical adsorption analyzer?
The static volumetric analysis instruments are diverse and have different characteristics for different application purposes, but all contain the following basic elements (pictured below): a vacuum pump, one or more gas sources, a metal or glass connected to the sample tube. Manifold, a coolant Dewar, a sample tube, a saturation pressure measuring tube, a pressure measuring device (pressure sensor). The volume of the manifold needs to be calibrated. There is a need for means to record manifold temperature. ? Sample tubes of various sizes can be used, and the volume is generally 10?~?20?cm3. To minimize the error, the freedom above the sample
The space should be as small as possible, and the dead volume can be reduced by placing a glass rod (filler rod) in the neck of the sample tube. ?
19. What are the requirements for physical purity of a physical adsorption analyzer?
Because the adsorbed gas is used to evaluate the surface area and pore size of the adsorbent, helium is used to determine the dead volume, so
According to ISO15901, the purity of these gases must be above 99.99%, but IUPAC pointed out in the latest report in 2015,
The purity of the adsorbed gas must not be lower than 99.999%. ?
20. Why do you want to weigh the sample (weighing)? How large is the sample size?
The specific surface area is the surface area per unit mass, so the sample in the sample tube must be weighed by weight reduction after degassing and before analysis. For nitrogen adsorption measurements, we consider the total surface area of ​​the sample in the sample tube, which is the specific surface area? X? Sample quality.
The sample amount is preferably between 5 and 200 200 m. What is measured by the balance is quality, not weight. For convenience of explanation, the two concepts are mixed here. A rough estimate of the accuracy of the instrument (the smallest total surface area measurable) and the surface area of ​​the sample should be made prior to testing to determine the amount of sample required. Less than the accuracy of the instrument, the relative error of the test results is larger. When the sample has a high specific surface area, the amount of the sample is small, and the weighing process may bring a large error. The principle of judging the amount of sample is:
1) Minimize the weighing error of the balance;
2) The sample in the sample tube should have sufficient adsorption capacity, and the pressure change caused by it should be much larger than that allowed by the pressure balance.
Tolerence;?
3) The amount of sample adsorption in the sample tube should not be too large, otherwise the adsorption equilibrium time is too long, resulting in too long experimental time;
4) If only specific surface area measurements are required, the amount of sample should be such that the total surface in the sample tube is at least between 1-5 μm 2 Å;
5) If the adsorption and desorption isotherm is determined, the total surface area in the sample tube should be at least 15-20 μm 2 . ??
For nitrogen adsorption, the experience with weighing is as follows:
-?
-?
Weigh as much as possible to 100mg or more to reduce weighing error?
If the specific surface is greater than 1000:??
Weighing 0.05?-0.08g?
If the specific surface is greater than 10, less than 1000: ? ? Weighing 0.1?-0.5g?
If the specific surface is less than 1:?
Weighing needs to be above 1g, even to 5g or more?
To ensure measurement accuracy, the quality of the sample should be reweighed after analysis. If the quality after analysis is not equal to after degassing, before analysis
The initial quality should be recalculated using the analyzed quality. ?
21. What are the specifications for the sample tube? What are the selection principles for sample tubes and filler rods?
General manufacturers are available in a variety of sample tubes with 6mm, 9mm and 12mm diameters. The finer the pipe diameter, the smaller the dead volume and the higher the measurement accuracy, but it is more difficult to load the sample. Therefore, according to the sample situation, weigh the pros and cons and use it as appropriate. ?
Reducing cold free space is a consensus among all instrument design and manufacturing personnel. Therefore, when selecting a sample tube, follow the principle of “use as much as possible with a packed rod, as thin a sample neck as possible, and as small as possibleâ€. ?
In addition, we need to consider the following factors:
1) Effect of sample morphology: For powder samples, especially low-density powder samples, such as activated carbon, the powder is lifted during vacuuming, which may cause inaccurate analysis results. If the powder is contaminated by O-rings, it will cause The system leaks, and once the powder enters the system manifold it will also cause more difficult system damage. Therefore, it is recommended to use tubes with relatively thick necks and relatively large sample compartments for such samples, and filler rods are not recommended. For large particles, high-density samples, such as metals, some molecular sieves, etc., are less affected by the vacuuming force and will not cause system pollution. Therefore, the selection of the sample tube can directly follow the first principle. Fine sample neck, as small as possible sample compartment." ?
2) Influence of type of analysis: For the pore size analysis of microporous materials, the molecular diffusion rate is slow at low temperatures due to the relatively low initial pressure of the experiment (usually starting from the relative pressure of 10-7/10-6 segments). Gas non-ideality has a large impact on data acquisition, so it is recommended not to use a filler rod to reduce experimental error. For the pore size analysis and specific surface area test of the mesoporous section, the effect of gas non-ideality on data acquisition is minimal, so the use of a packed rod can improve the accuracy of the experimental results. ?
3) Effect of sample specific surface area: For small specific surface samples, the amount of sample required during the test is large, usually requiring several grams or even ten grams. In this case, to ensure the accuracy of the experiment, it should be noted that the sample volume should not exceed 2/3 of the total volume of the sample chamber (straight tube, small ball or large ball). In addition, if the small surface sample also has the characteristics of low density mentioned in 1), then
A filler rod is recommended. ?
The selection experience of the sample tube has the following reference:
ï¬ 9?mm? The sample tube is the most commonly used sample tube and is suitable for most samples;
ï¬ Standard sample tubes for particle samples and conventional specific surface analysis;
ï¬ Large ball sample tube for powder samples and low surface sample analysis;
ï¬ 6mm sample tube is necessary for high precision microwell analysis. ?
?
22. What is a manifold? How does it affect the accuracy of the instrument?
?
The manifold is a multi-branch piping system that connects the intake port, vacuum system, pressure sensor, and sample tube in a physical adsorption analyzer. Manifold volume is one of the basis for calculating the initial intake of physical adsorption. This part of the volume is solidified inside the instrument and can be corrected to obtain accurate values. On the other hand, the greater the pressure difference of the adsorbate gas during the diffusion process, the more accurate the absolute amount of gas is calculated. Therefore, the smaller the manifold volume, the higher the accuracy of the instrument. ?
23. Why record the manifold temperature? What is the effect of manifold temperature control on measurement accuracy?
In the ideal gas equation, volume and pressure are functions of temperature. Therefore, accurate system temperature is also a basis for accurate calculation of adsorption capacity. Typically the system temperature is recorded in real time through a temperature sensor connected to the manifold. Most of the instruments currently on the market use temperature sensors with an accuracy of ±0.1 ° °C, which can meet the requirements of experimental accuracy. However, it must be pointed out that the latest instrument design trend is the so-called "high-resolution micropore analysis" technology, which uses a 0.1?torr pressure sensor to collect low-pressure zone data so that it is in a high vacuum region (relative pressure <10) -6) The data resolution and stability are higher. However, this type of sensor is more sensitive to temperature changes. Therefore, in order to obtain high data stability, it is necessary to specially configure a more stable system temperature, for example, by means of system heating, keeping the manifold at 50 ° C, avoiding temperature. fluctuation. ?
If it is a static high-pressure adsorption system, the manifold temperature fluctuates by ±0.5 ° °C, which will cause a significant error in the calculation of the adsorption amount (such as ±0.3?mol?@?CO2). Therefore, the temperature control accuracy of the manifold temperature is required to be ± Within 0.1? °C. ?
The pressure sensor, as the basic metering unit of the static capacity method, should have its own electronic ceramic thermostat system. If a pressure sensor without a thermostat is used, although the cost is low, the pressure measurement accuracy is extremely low, and it is impossible to measure a large mesoporous distribution of 10 nm or more. ?
24. Why do you want to get rid of helium before the analysis process begins?
The measurement of dead volume with helium is based on the assumption that helium is not adsorbed. But in fact, physical adsorption is non-specific adsorption, and there is adsorption on any gas. Therefore, some materials, especially microporous materials, will adsorb more helium, and its influence cannot be ignored.
That is, there is æ°¦ pollution. A typical phenomenon of æ°¦ contamination is that the adsorption isotherm appears as an "S" line shape when P/P0 < 10-5 or less. Therefore, in this case, it should be concerned whether the helium removal process has been performed after the dead volume measurement, and then the isotherm measurement is performed; or after the adsorption isotherm is measured, it is corrected. The IUPAC report in the 2015 report states that recent studies have confirmed that nanoporous solids with extremely narrow micropores can absorb unimportant amounts of helium (æ°¦ interception) at liquid nitrogen temperatures. If the trapped helium is not removed before the analysis, it can significantly affect
The shape of the adsorption isotherm in the ultra-low pressure range. Therefore, it is recommended that at least the sample should be degassed after it has been allowed to overflow at room temperature before continuing the analysis. As a monoatomic molecule, helium is only 0.26 nm in diameter, much smaller than the cross-sectional area of ​​nitrogen molecules, and can enter the extremely fine pores where nitrogen cannot enter. Studies have shown that the BET specific surface of activated carbon fibers is analyzed by helium at liquid helium temperature.
Characterized by nitrogen at liquid nitrogen temperature, the BET specific surface area value is increased by 1/3. ?
25. What is cold free space? What is warm free space? What is the significance of the relative size of the free space? ?
During the analysis, the sample tube is partially immersed in a cold bath (such as liquid nitrogen), so the total free space is composed of cold free space and warm free space. The part immersed in the liquid nitrogen level is called cold free space (cold-zone), and the part at room temperature above the liquid level is called warm-space (warm-zone). Free space is the area where the adsorbate molecules in the system transmit and diffuse. At the liquid nitrogen temperature (77?K), the number of molecules contained in the same volume is 4 times that of 300?K at room temperature. It can be said that the contribution of cold free space to dead volume in the whole free space is much larger than that of warm free space. The smaller the cold free space, or the more gas molecules contained in the warm free space, the more accurate the pressure measurement. ?
26. Why do you want to control the liquid level of liquid nitrogen or liquid argon? What are the methods for controlling the liquid level?
In an open Dewar, refrigerants such as liquid nitrogen and liquid argon are volatilized. As a result, the refrigerant level of the sample neck is continuously reduced, resulting in a continuous change in the volume of the cold and warm regions. In order to avoid the system free space affected by the cold bath temperature and the cold bath level,
The key to the measurement is to maintain a relatively constant sample neck and refrigerant level. Generally, at least 20mm of the sample is immersed, and the liquid level is kept constant, and the fluctuation does not exceed 1~2mm. In practice, there are two different ways to achieve this:
(1) RTD real-time feedback servo mode, which uses a real-time feedback servo system including liquid level sensor and automatic elevator to adjust the cold bath level and keep the cold free space to a minimum. Experiments have shown that the level of the sample cell immersed in liquid nitrogen is controlled by a liquid level sensor, and the nitrogen pressure (about 295 mm Hg) in the sample tube is shown as a function of time. Any change in the liquid nitrogen level will cause a change in the pressure inside the tube. The constant pressure results clearly show that the liquid level control servo feedback system compensates for the loss of liquid nitrogen evaporation and excellently controls the dead volume (minimum "cold zone" and maximum "warm zone") in the sample tube to remain constant . ?
(2) Jacket method, that is, the sample neck is wrapped with a porous polymer material, and the liquid level is maintained by capillary transpiration, that is, the temperature of the cold bath is constant by using a large cold free space. ?
27. What is saturated vapor pressure? Why measure saturated vapor pressure?
• The vapor of the substance is present on the surface of the liquid (or solid). The pressure generated by the vapor on the surface of the liquid (or solid) is called the vapor pressure of the liquid (or solid). The pressure generated by steam at equilibrium with the liquid (or solid) state of the same substance at a certain temperature
Strong is the saturated vapor pressure. ? ? ? ? When the sample tube is immersed in a refrigerant (such as liquid nitrogen), the saturated equilibrium vapor pressure of the pure adsorbate (nitrogen) in the sample tube is represented by P0. When the liquid pure material vapor has a pressure of its saturated vapor pressure, the vapor-liquid two phases reach a phase equilibrium. Therefore, under vacuum saturation adsorption, the adsorption pressure cannot be greater than the saturated vapor pressure of the adsorbed material, that is, the relative pressure (P/P 0) cannot be greater than 1. ?? ? ? Saturated vapor pressure is an important property of the adsorbed material, its size depends on the nature and temperature of the material, and the thickness of the adsorbed layer,
The pore filling pressure is related to capillary condensation in the pores. • The greater the saturated vapor pressure, the more volatile the material is. Only by obtaining accurate gas saturated vapor pressure, accurate pore size and specific surface area analysis can be performed by accurate characterization of the relationship between adsorption amount and relative pressure P/P0. ?
28. How to measure saturated vapor pressure?
The magnitude of the saturated vapor pressure is temperature dependent. There are a variety of experimental methods that can be used to calculate the saturated vapor pressure during physical adsorption. However, the most accurate method is to continuously measure the saturated vapor pressure in a separate P0 tube during the physical adsorption experiment. Usually, the adsorption isotherms are measured at liquid nitrogen (77.35?K) or liquid argon (87.27?K?). Liquid nitrogen and liquid argon are placed in Dewar to maintain normal pressure. At this time, the liquid temperature is not only related to pressure but also to liquid purity. Water vapor, oxygen and other gas components in the air can affect the purity of the liquid. When the purity of the liquid decreases, the temperature of the liquid will also increase. The temperature rise of 0.1?~?0.2?K can cause the saturated vapor pressure to rise. ?~?20?torr. In the physical adsorption process, when the relative pressure is 0.95, the error of the saturated vapor pressure reaches 5? torr, which will cause nearly 10% error in the aperture calculation. Therefore, it is very important to measure the saturated vapor pressure accurately and in real time during the physical adsorption process. ?
An instrument with an independent saturated vapor pressure sensor can monitor the change of P0 in real time without interfering with the experimental process. However, if the saturated vapor pressure in the relative pressure is not taken from the saturated vapor pressure at the equilibrium time of the point, the accuracy of the measurement will still be greatly discounted, which is of great significance for the analysis of the micropore distribution of the microporous material. ?
29. 物ç†å¸é™„分æžç³»ç»Ÿçš„进气模å¼éƒ½æœ‰å“ªäº›ï¼Ÿå„有什么特点?
由于物ç†å¸é™„分æžç³»ç»Ÿæµ‹å®šçš„基础数æ®æ˜¯å¹³è¡¡å¸é™„é‡ä¸ŽåŽ‹åŠ›çš„å…³ç³»ï¼Œå› æ¤æˆ‘们必须设定一个é‡å€¼ï¼Œè€Œæµ‹å®šå¦ä¸€ä¸ªé‡å€¼ã€‚è¿™æ ·ï¼Œå°±äº§ç”Ÿäº†ä¸¤ç§è¿›æ°”模å¼ï¼š?
(1) 定投气é‡æ¨¡å¼ï¼ˆè®¾å®šçºµåæ ‡ï¼Œæµ‹é‡æ¨ªåæ ‡ï¼‰ï¼š?
由仪器采集压力信æ¯çš„方法称之为“定投气é‡æ–¹å¼â€ã€‚该方法对于仪器硬件åŠå›ºä»¶è®¾è®¡çš„è¦æ±‚较低,是å„个生产厂家广泛使用的方法。该方法的一个亮点是å¯ä»¥æ‰©å±•è¿›è¡Œå¸é™„动力å¦çš„ç›¸å…³ç ”ç©¶ä»¥åŠä½Žæ¸©ååº”çš„ç›¸å…³ç ”ç©¶ï¼Œä½†å¯¹äºŽå¸¸è§„çš„å¾®å”å”径分布分æžï¼Œå®šæŠ•æ°”é‡æ–¹å¼å˜åœ¨å¦‚下ä¸ç¡®å®šæ€§ï¼š? ?如果投气é‡è®¾ç½®è¿‡å°ï¼Œå¾—到的ç‰æ¸©çº¿å›ºç„¶ç»†èŠ‚丰富,但是å´ä¸Žå®žéªŒæ‰€èŠ±æ—¶é—´å‘ˆå比。如果投气é‡è®¾ç½®å大,ç‰æ¸©çº¿ä¸Šçš„部分信æ¯å°±ä¼šä¸¢å¤±ã€‚?投气é‡è®¾ç½®å大,å¯ä»¥ç¼©çŸæµ‹è¯•æ—¶é—´ï¼Œä½†å¹¶æ²¡æœ‰è¾¾åˆ°çœŸæ£çš„å¸é™„å¹³è¡¡ï¼Œé€ æˆå¸é™„ç‰æ¸©çº¿å‘å³â€œæ¼‚
移â€ï¼Œå¯¼è‡´å¾®å”分æžçš„误差(è§å›¾49-1)。?
图49-1 以定投气é‡çš„æ–¹å¼ï¼Œç”¨ä¸åŒçš„投气
图49-2 以定投气é‡çš„æ–¹å¼ï¼Œç”¨ä¸åŒçš„平衡
? 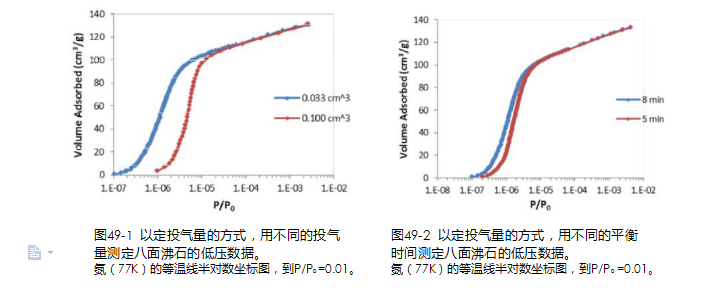
é‡æµ‹å®šå…«é¢æ²¸çŸ³çš„低压数æ®ã€‚
氮(77K)的ç‰æ¸©çº¿åŠå¯¹æ•°åæ ‡å›¾ï¼Œåˆ°P/P0 =0.01。时间测定八é¢æ²¸çŸ³çš„低压数æ®ã€‚氮(77K)的ç‰æ¸©çº¿åŠå¯¹æ•°åæ ‡å›¾ï¼Œåˆ°P/P0 =0.01。IUPAC 在2015 年的报告ä¸æŒ‡å‡ºï¼šå¤ªçŸçš„平衡时间会导致未平衡的数æ®ç”Ÿæˆï¼Œç‰æ¸©å¸é™„线移å‘è¿‡é«˜çš„ç›¸å¯¹åŽ‹åŠ›ã€‚å› ä¸ºåœ¨çª„å¾®å”ä¸çš„平衡往往是éžå¸¸æ…¢çš„,未平衡往往是在ç‰æ¸©çº¿çš„æžä½Žç›¸å¯¹åŽ‹åŠ›åŒºåŸŸå†…容易å‘生的问题(è§å›¾49-2)。? ?
(2) 定压力方å¼ï¼ˆè®¾å®šæ¨ªåæ ‡ï¼Œæµ‹é‡çºµåæ ‡ï¼‰ï¼š?
由仪器采集并计算饱和å¸é™„é‡çš„方法称之为“定压力方å¼â€ï¼Œè¯¥æ–¹æ³•æœ€å¤§çš„优点是:由仪器内置程åºè®¡ç®—å„定义压力下的å¸é™„é‡ï¼Œè¿™ç§æ–¹æ³•å¯¹äºŽå¸é™„é‡æœªçŸ¥çš„æ ·å“å¯ä»¥æ—¢å¿«åˆå‡†åœ°å¾—到å¸é™„ç‰æ¸©çº¿ï¼Œ
尤其对于未知的微å”æ ·å“。 ?快速ã€å‡†ç¡®åœ°æµ‹é‡ä¸Žæ•°æ®çš„准确性åŒæ ·å…·æœ‰é‡è¦å®žè·µæ„义。但是,定压力方å¼å¯¹å†…置程åºè®¾è®¡è¦æ±‚æžé«˜ï¼Œå°¤å…¶æ˜¯å¯¹äºŽå¾®å”定压力测é‡ï¼ˆå®žéªŒèµ·å§‹ç›¸å¯¹åŽ‹åŠ›éœ€è¾¾åˆ°10-7~10-5é‡çº§ï¼‰ï¼Œå¿…é¡»åŒæ—¶è€ƒè™‘饱和蒸汽压ã€ç³»ç»Ÿä½“积ã€æ ·å“é‡ç‰ä¿¡æ¯ï¼Œå…·æœ‰å…¶å¤æ‚性。ä¸æ£ç¡®çš„“定压力方å¼â€å®å‘½ä»¤ç¼–程设计很容易导致ç‰æ¸©çº¿æµ‹é‡çš„å差。 ?
30. å¸é™„平衡æ¡ä»¶æ˜¯å¦‚何设置的?
在é™æ€å®¹é‡æ³•ç‰©ç†å¸é™„实验ä¸ï¼Œæ‰€è°“å¸é™„平衡是在一定的扩散时间内,体系ä¸æ°”体压力å˜åŒ–始终在å…许误差范围内的状æ€ã€‚它与投气方å¼å…±åŒç»„æˆäº†ç‰©ç†å¸é™„仪器测é‡å‡†ç¡®åº¦ä¸æœ€æ ¸å¿ƒçš„环节。 ?若平衡时间ä¸å¤Ÿï¼Œåˆ™æ‰€æµ‹å¾—çš„æ ·å“å¸é™„é‡æˆ–脱附é‡å°äºŽè¾¾åˆ°å¹³è¡¡çŠ¶æ€çš„é‡ï¼Œè€Œä¸”å‰ä¸€ç‚¹çš„ä¸å®Œå…¨å¹³è¡¡è¿˜ä¼šå½±å“到åŽé¢ç‚¹çš„测定。例如,测定å¸é™„曲线时,在较低相对压力没有完æˆçš„å¸é™„é‡å°†åœ¨è¾ƒé«˜çš„压力点被å¸é™„,这导致ç‰æ¸©å¸é™„线å‘高压方å‘ä½ç§»ã€‚由于åŒæ ·çš„å½±å“,脱附曲线则å‘低压方å‘ä½ç§»ï¼Œå½¢æˆåŠ 宽的回滞环,或者产生ä¸å˜åœ¨çš„回滞环。 ? 对于微å”测é‡ï¼Œç”±äºŽå…¶å”径较å°ï¼Œéœ€è¦çš„å¹³è¡¡æ—¶é—´ç›¸åº”å¢žåŠ ã€‚ ?
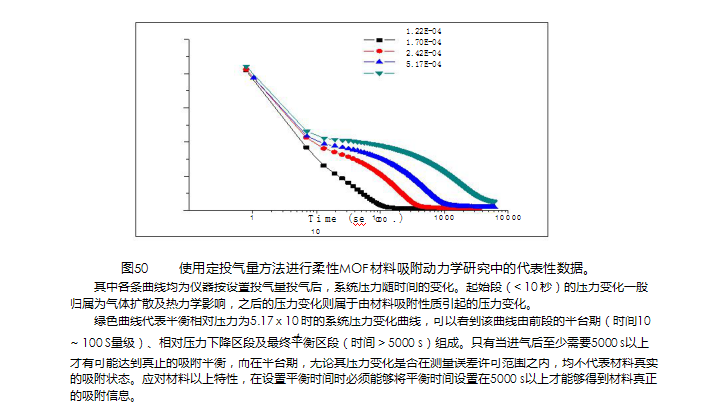
使用定投气é‡æ–¹æ³•è¿›è¡ŒæŸ”性MOFææ–™å¸é™„动力å¦ç ”究ä¸çš„代表性数æ®ã€‚å…¶ä¸å„æ¡æ›²çº¿å‡ä¸ºä»ªå™¨æŒ‰è®¾ç½®æŠ•æ°”é‡æŠ•æ°”åŽï¼Œç³»ç»ŸåŽ‹åŠ›éšæ—¶é—´çš„å˜åŒ–。起始段(< 10 秒)的压力å˜åŒ–一般归属为气体扩散åŠçƒåŠ›å¦å½±å“,之åŽçš„压力å˜åŒ–则属于由ææ–™å¸é™„性质引起的压力å˜åŒ–。绿色曲线代表平衡相对压力为5.17 x 10 时的系统压力å˜åŒ–曲线,å¯ä»¥çœ‹åˆ°è¯¥æ›²çº¿ç”±å‰æ®µçš„å¹³å°æœŸï¼ˆæ—¶é—´10-4~ 100 Sé‡çº§ï¼‰ã€ç›¸å¯¹åŽ‹åŠ›ä¸‹é™åŒºæ®µåŠæœ€ç»ˆå¹³è¡¡åŒºæ®µï¼ˆæ—¶é—´> 5000 s)组æˆã€‚åªæœ‰å½“进气åŽè‡³å°‘需è¦5000 s以上æ‰æœ‰å¯èƒ½è¾¾åˆ°çœŸæ£çš„å¸é™„平衡,而在平å°æœŸï¼Œæ— 论其压力å˜åŒ–是å¦åœ¨æµ‹é‡è¯¯å·®è®¸å¯èŒƒå›´ä¹‹å†…,å‡ä¸ä»£è¡¨æ料真实的å¸é™„状æ€ã€‚应对æ料以上特性,在设置平衡时间时必须能够将平衡时间设置在5000 s以上æ‰èƒ½å¤Ÿå¾—到æ料真æ£çš„å¸é™„ä¿¡æ¯ã€‚如果说平衡时间(Equilibrium?time)规定的是达到平衡的最低时间è¦æ±‚,那么平衡压力误差
(Tolerance)则是用于认定达到平衡时å…许压力å˜åŒ–范围的å‚数。 ?这两个å‚æ•°å…±åŒå†³å®šäº†å¸é™„平衡æ¡ä»¶ã€‚éšç€å„ç§ç‰¹è‰²æ–°æ料的快速涌现,å¸é™„平衡æ¡ä»¶è®¾ç½®å¿…须具有足够的çµæ´»æ€§ä»¥é€‚应ä¸åŒç±»åž‹æ料分æžçš„需求。例如,对于柔性MOFæ料(也有人称之为会呼å¸çš„æ料),由于其å”é“结构å˜åŒ–需è¦ç›¸å½“长的时间,在实验平衡æ¡ä»¶è®¾ç½®æ—¶ï¼Œå¿…须能够针对具体æ料的å”é“结构å˜åŒ–时间设定仪器的平衡时间(è§å›¾50)。 ?
å› æ¤ï¼Œèƒ½å¦è¿›è¡Œçµæ´»çš„å¸é™„平衡æ¡ä»¶è®¾ç½®å°±æˆäº†è¡¡é‡ç‰©ç†å¸é™„仪器测é‡å‡†ç¡®åº¦çš„一个é‡è¦æ ‡å‡†,?
According to the needs of different office environments, office projectors are roughly divided into three categories.
One is a conventional projector that is placed or hoisted in the conference room, the second is a Portable Projector that can be carried around, and the third is an ultra-short-throw projector for convenient work reports and speeches.
With the advent of the Internet era, the emergence of wireless office series represented by ViewSonic PJD6352, PJD6550LW, PJD6252L, and PJD6552LWS has added new members to the office projectors. The wireless Office Projector is realized by the built-in wireless module of the ordinary office projector. Wireless office enables people not to switch signal lines frequently during office meetings, allowing people to have a better experience in meetings.
4k meeting room projector,video projector for meeting room,good projector for meeting room,conference room led projector,lcd meeting room projector
Shenzhen Happybate Trading Co.,LTD , https://www.happybateprojector.com